|
| Dr Sergio Martinez Cuesta, Dr Tim Stone and Dr Dean Hammond |
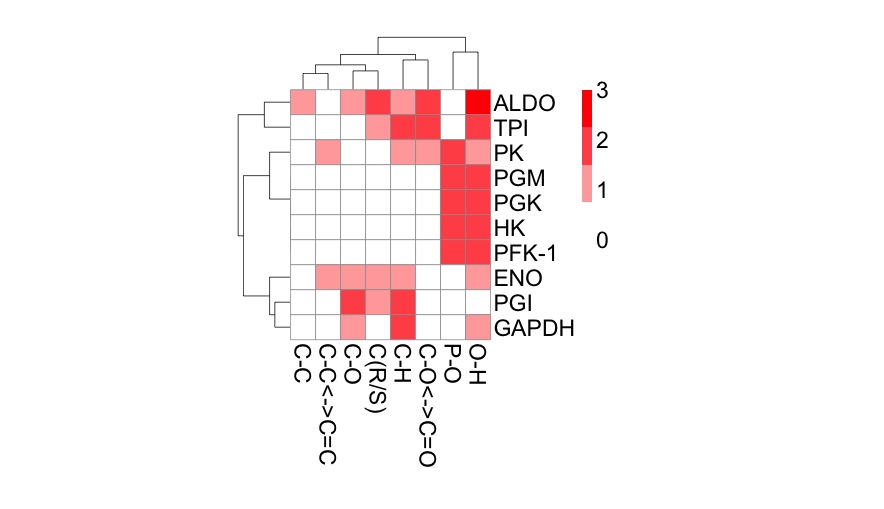
I learned about principal component analysis, microarray analysis and getting data into R can be quite challenging. I also learned that you can import pictures files into R. I'll write more about that in the future.
I hope the attendees enjoyed it and learned something. They generally seemed happy with the day but I will have to await the formal feedback.
For those that attended and others, here are a few resources that might help you to keep learning:
- The Quick R website
- The R Graphics Cookbook site - lots about ggplot
- Biostars.org - a bioinformatics help website
- StackOverFlow - most of your questions have been answered but you can always ask
- R-bloggers - a treasure trove of information about R with more added everyday.
However, the most important things to be are diligent (spend a little time each day playing with R) and patient (things take time to learn and longer to master).