Here is a coloured kinome tree that I am kind of happy with for the moment. I think there is some improving still to do but it's nicer than the one coloured version I made before.

The code below uses a very manual approach to manipulating trees. There are various reasons for that and these include the fact that I want to make an unrooted tree. There are ways to manipulate trees using other packages including phytools and ggtree. However, I'm not able to get them to work well with unrooted trees. There is still more to learn....
Here is the code that I used to draw this and some of the trees that I made along the way:
START
library(ape)
library(RCurl)
# a tree was made using code in a previous blog post.
# see here: http://rforbiochemists.blogspot.co.uk/2017/01/visualizing-kinome-in-r-simple-tree.html
# it's here on github
link <- "https://raw.githubusercontent.com/brennanpincardiff/RforBiochemists/master/phyloTrees/kinaseTree_20161221"
# this will download it into R.
# using read.tree() from ape package
tree <- read.tree(file = link)
# this tree looks quite nice in my opinion and is the starting point of this blog
plot(tree, "u",
use.edge.length = FALSE,
show.tip.label = FALSE,
edge.color = "red")
# but lacking colours for the groups and any labels...

# it seems more traditional to draw trees from left to right.
plot(tree,
use.edge.length = FALSE,
show.tip.label = FALSE,
edge.color = "red")

# with some names... this is slow to draw due to the names
plot(tree,
use.edge.length = FALSE,
edge.color = "red",
cex = 0.25)

# to customise this tree in a way we want we need to understand a little more about trees
# we can find out more about an object by writing the name
tree
# "Phylogenetic tree with 516 tips and 514 internal nodes"
# by using the class() function
class(tree)
# "phylo"
# or by using the str() structure function
str(tree)
# "List of 4"
# this list includes $edge, $Nnode, $ tip.label and $edge.length
# the tree$tip.label includes family designation
tree$tip.label # 516 of these
# from the Science paper, we have seven kinase families:
# kinase categories... TK, TKL, STE, CK1, AGC, CAMK, CMGC
# with the following colours
# "red", "green", "paleblue", "orange", "yellow", "purple", "pink", "green"
# by using the grep()function on the tree$tip.label part of the object
# we can find the tip labels that include "TK/" - i.e. tyrosine kinases
grep("TK/", tree$tip.label) # gives a list of numbers with "TK/" in tip label
length(grep("TK/", tree$tip.label))
# thus there are 94 tip labels with that are designated TK (not TKL tyrosine kinase like)
# make a vector for each tip.label called tipcol with black on all of these...
tipcol <- rep('black', length(tree$tip.label))
# make a vector with our list of kinase categories
kinaseCats <- c("TK/", "TKL", "STE", "CK1", "AGC", "CAMK", "CMGC", "RGC")
# make a vector of color we want:
colorsList <-c("red", "darkolivegreen3", "blue", "orange", "yellow", "purple", "pink", "green")
# replace colours where grep gives "TK" as red, etc in a loop
for(i in 1:length(kinaseCats)){
tipcol[grep(kinaseCats[i], tree$tip.label)] <- colorsList[i]
}
# plot with edge length false to see nodes better
plot(tree,
use.edge.length = FALSE,
tip.color=tipcol,
cex = 0.25)
# slow to draw due to text - a bit annoying!
 |
| Kinome tree with different coloured labels for different kinds of kinases. Tyrosine kinases are in red. |
# trees are made up of nodes and edges.
# its possible to label nodes using nodelabels() function from ape package
nodelabels(cex=0.4)
# labels internal nodes.
 |
| Internal nodes of the tree are labelled with the number |
# the only way seems to identify the relevant nodes manually
# i.e. the nodes that include the kinase groups that we have coloured
# from the bottom
# for 1st "green" looks like node 574
# for "red" looks like node 607
# for 2nd "green" somethink like 701 but very difficult to see
# for "purple" most of 749 and also north of 726 but I can't read the number
# "blue" node 723
# "yellow" node 885, I think
# "pink" node 955, I think
# not perfect but getting there....
# adding edge colors
# from 111 to 177 should be green
# from 178 to 364 should be red.
# from 459 to 577 should be purple
# from 578 to 608 should be purple too
# from 641 to 733 should be blue
# from 735 to 850 approx should be yellow
# from 876 to 980 should be pink
# http://stackoverflow.com/questions/34089242/phylogenetic-tree-tip-color
# make a vector for each edge called edgecol with black on all of these...
edgecol <- rep('black', nrow(tree$edge))
edgecol[178:364] <- "red" # "TK/"
edgecol[111:177] <- "green" # "TKL" OR "RGC"
edgecol[641:733] <- "blue" # "STE"
edgecol[1003:1029] <- "orange" # "CK1"
edgecol[735:850] <- "yellow" # "AGC"
edgecol[459:577] <- "purple" # "CAMK"
edgecol[578:608] <- "purple" # "CAMK"
edgecol[876:980] <- "pink" # "CMGC"
plot(tree,
use.edge.length = FALSE,
tip.color=tipcol,
edge.color = edgecol,
cex = 0.25)
 |
| Kinome Tree with text and branches coloured. |
plot(tree, "u",
use.edge.length = FALSE,
tip.color=tipcol,
edge.color = edgecol,
cex = 0.25)

# plot.phylo() function from ape package allows rotation of tree.
plot.phylo(tree, "u", use.edge.length = FALSE,
edge.color = edgecol,
rotate.tree = -95,
show.tip.label = FALSE)

# want to add some names at the ends of the branches
# try to find out some tip numbers using
tiplabels(cex=0.3)

# add labels to node 246, 105, 191 and 340
tree$tip.label[246] # "CaMK1d_Hsap_-CAMK/CAMK1"
tree$tip.label[105] # "EphA3_Hsap_-TK/Eph"
tree$tip.label[191] # "TGFbR1_Hsap_-TKL/STKR/Type1"
tree$tip.label[340] # "TNIK_Hsap_-STE/STE20/MSN"
# add these to a list
kinaseLabels <- c("IKKa","ErbB2", "MLK1", "PKCb",
"CDK9", "CaMK1d_Hsap_-CAMK/CAMK1",
"EphA3_Hsap_-TK/Eph", "TGFbR1_Hsap_-TKL/STKR/Type1",
"TNIK_Hsap_-STE/STE20/MSN")
# extract tip.labels
tipLabels <- tree$tip.label
# find these in the alignment - they will be tip labels
labelNo <- NULL
for(i in 1:length(kinaseLabels)){
labelNo <- c(labelNo, grep(kinaseLabels[i], tree$tip.label))
}
# generates a vector of 9 numbers. Some labels in two names.
# make a vector of blank tiplabels
tipLabels_2 <- rep('', length(tree$tip.label))
# add the labels we want to the vector...
for(i in 1:length(labelNo)){
tipLabels_2[labelNo[i]] <- tipLabels[labelNo[i]]
}
# make a new tree
tree_fewLabels <- tree
# replace the tip labels with the shorter list.
tree_fewLabels$tip.label <- tipLabels_2
# remove "Hsap"
tipLabels_2 <- gsub("Hsap", "", tipLabels_2)
tree_fewLabels$tip.label <- tipLabels_2
# plot.phylo() function from ape package allows rotation of tree.
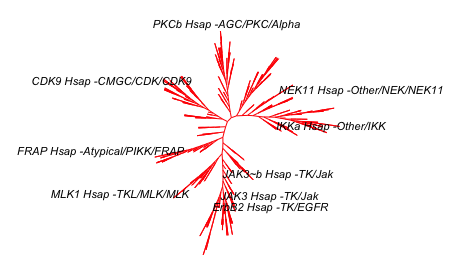
plot.phylo(tree_fewLabels, "u",
use.edge.length = FALSE,
edge.color = edgecol,
rotate.tree = -95,
show.tip.label = TRUE,
cex = 0.4)

# add a title and source
plot.phylo(tree_fewLabels, "u",
main="Phylogenetic tree of human kinase domains",
sub="source: www.kinase.com & Manning et al Science (2002) 398:1912-1934",
rotate.tree = -95,
use.edge.length = FALSE,
edge.color = edgecol,
show.tip.label = TRUE, font = 2,
cex = 0.5)

# this looks quite good and is enough for today.