



Update: 1 July 2025 - so a lot has happened in 10 years. This includes people moving jobs, promotions and the reorganisation of data on published website.
Aled was promoted to Professor at Cardiff University
----
I have chosen a paper from Molecular and Cellular Proteomics which uses aptamers to detect multiple proteins in exosomes and cells.
The data was generated by colleagues that were working in the School of Medicine at Cardiff University including the first author - Dr Jason Webber, a Prostate Cancer UK funded Research Fellow and senior author, Professor Aled Clayton, a Senior Lecturer in Cancer & Genetics at Velindre Hospital. Dr Tim Stone was key to the data analysis. The protein detection method is from a company called SomaLogic.
The first step was downloading the file from the Molecular and Cellular Proteomics website. I used the download.file() function. This saves the file into your current directory. This was opened using the readxl package (by Hadley Wickham) using the read_excel() function.
I drew some graphs as I explored and manipulated the data. These are interspersed with the script below. The visualisations included boxplots and a cluster diagram.
I wanted to draw a volcano plot which expresses the fold change against the significant of the change (p-value). I couldn't do that with the data supplied so I had to reverse the transformation and calculate the fold change again.
Here is the volcano plot:
 |
| Comparison of changes in exosomes compared cells. Proteins over-expressed in exosomes are on the right. Proteins over-expressed in cells are on the left. |
The plot indicates that there are more proteins over-expressed in cells (on the left) compared to exosomes (on the right).
Update: 1 July 2025 - orignally, I was able to download the data directly from the Molecular and Cellular Proteomics website. However, at some point, they reorganised their site and put all the data into a zip file. This means that downloading it requires multiple steps outside of R. To make this analysis more stand alone, I have down loaded the data from this zip file and uploaded the spreadsheet onto my Github site. This means that the script will download and analyse the data.
Here is the script with some other plots along the way:
START
# pull down a file from the internet, do some analysis and draw a graph...
# choose Jason Webber's MCP Paper...
# the data can be downloaded using this link with give zip file.
# It isn't necessary to do that for this script.
# install if necessary:
# install.packages(c("ggplot2", "readxl"))
library(ggplot2)
library(readxl)
# this is the link to the data
link <- "https://github.com/brennanpincardiff/RforBiochemists/raw/master/R_for_Biochemists_101/data/mcp.M113.032136-6.xlsx"
# the download.file() function downloads and saves the file with the name given
download.file(url=link,destfile="file.xlsx", mode="wb")
# then we can open the file and extract the data using the read_excel() function.
data<- read_excel("file.xlsx")
View(data)
# plot the data - always an important first step!
boxplot(data[2:7],
las =2, # las = 2 turns the text around to show sample names
ylab = expression(bold("expression")),
main="Boxplot of expression data")
 |
| Two types of sample - exosomes (n=3) and cells (n=3). |
# do a cluster analysis to quality control the different groups
# convert to matrix first
data.m <- as.matrix(data[2:7])
dim(data.m) # gives the dimensions of the matrix
# ans: 762 6
# calculate the distances using the dist() function.
# various methods are possible - default is Euclidean.
distances <- dist(data.m)
summary(distances) # have a look at the object
# make the cluster dendrogram object using the hclust() function
hc <- hclust(distances)
# plot the cluster diagram
# some interesting groups in the data
plot(hc,
xlab =expression(bold("All Samples")),
ylab = expression(bold("Distance")))
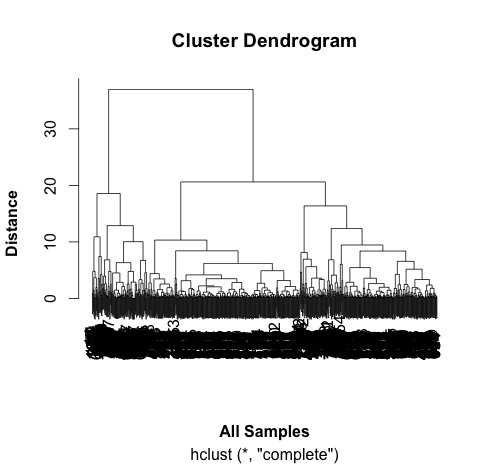
# ah, not what I intended.
# it clustered the proteins NOT the samples.
# transpose the data and try again...
data.m.t <- t(data.m)
dim(data.m.t)
# ans = 6 762 - so that has worked.
# repeat cluter analysis
# calculate the distances using the dist() function.
# various methods are possible - default is Euclidean.
distances <- dist(data.m.t)
summary(distances)
# make the cluster dendrogram object using the hclust() function
hc <- hclust(distances)
# plot the cluster diagram
# some interesting groups in the data
plot(hc,
xlab =expression(bold("All Samples")),
ylab = expression(bold("Distance")))
# replicates cluster together well.

# the adjusted P value are in a column entiteld: BH - P.value
# this is a little awkward so rename this and Fold Change column:
colnames(data)[9] <- "P.Value"
colnames(data)[10] <- "Fold.Change"
plot(data$P.Value ~ data$Fold.Change)

# this works but we would like to turn it into a volcano plot
# with log2 at the bottom and -p-value.
# we can't log fold changes as half of them are negative numbers
# we need to re-calculate the raw data
# reverse the log2 transformation.
# mean the values & calculate fold change in decimal format (no +/-)
data$exo1 <- 2^data$exoRFU1
data$exo2 <- 2^data$exoRFU2
data$exo3 <- 2^data$exoRFU3
data$cell1 <- 2^data$cellRFU1
data$cell2 <- 2^data$cellRFU2
data$cell3 <- 2^data$cellRFU3
# calculate means of the replicates
data$exoMean <- rowMeans(data[,13:15])
data$cellMean <- rowMeans(data[,16:18])
# always good to visualise the data:
plot(log2(data$exoMean)~log2(data$cellMean))

# calculate fold change exo/cell
data$FoldChange <- data$exoMean/data$cellMean
plot(log2(data$FoldChange)) # transform for plotting

# make column in data.frame with transformed data
data$Log2.Fold.Change <- log2(data$FoldChange)
## Identify the genes that have a p-value < 0.05
data$threshold = as.factor(data$P.Value < 0.05)
## Construct the volcano plot object using ggplot
g <- ggplot(data=data,
aes(x=Log2.Fold.Change, y =-log10(P.Value),
colour=threshold)) +
geom_point(alpha=0.4, size=1.75) +
xlim(c(-6, 6)) +
xlab("log2 fold change") + ylab("-log10 p-value") +
theme_bw() +
theme(legend.position="none") +
ggtitle("Volcano Plot comparing protein expression in exosomes vs cells ") # add a title
g # show the plot
END of script
So I think that the threshold of p<0.05 is too low for this volcano plot. It's relatively easy to change and would make a good exercise. Perhaps a threshold of p<0.00001 would be better.
Useful resources (updated 1 July 2025)
- Here is a script about making multiple volcano plots
- More information about volcano plots on Wikipedia.
- This Enhanced Volcano package looks interesting but I haven't played with it myself.